
Pharmacovigilance is an essential component of the healthcare industry that plays a crucial role in ensuring the appropriate benefit-risk balance of medicinal products. The pharmacovigilance system is a set of processes and procedures that are designed to detect, assess, and prevent adverse drug reactions and other safety concerns associated with pharmaceutical products.
The importance of pharmacovigilance cannot be overstated. Pharmaceutical products are developed to improve patients’ health and well-being but can also have unintended consequences. Therefore, it is essential to have a robust pharmacovigilance system in place to identify any safety concerns and take appropriate action to protect public health. Following you will find a short overview of the main components of PV system.
Quality System
Quality systems (QMS) are the set of procedures, policies, and practices that a company uses to ensure that its products or services meet or exceed customer expectations. A robust quality system is critical for ensuring consistent product or service quality, regulatory compliance, and customer satisfaction.
The main goal of QMS is to have oversight of how the system works. A good QMS shall have mechanisms/processes in place to identify and mitigate risks that could impact product quality or customer satisfaction. By following established SOPs, employees can proactively identify potential quality, and process issues and take corrective action before they become significant problems.
Quality systems should also be designed to promote continuous improvement. SOPs are critical for this process because they are cornerstone for process understanding and performance measurement. By tracking performance against established SOPs as well as legal regulations, businesses can identify areas for improvement and adjust their processes to improve quality.
SOPs (Standard Operating Procedures)
In today’s fast-paced and constantly evolving business environment, maintaining high-quality standards is paramount. Standard Operating Procedures (SOPs) are essential to quality systems that help businesses achieve consistent and reliable performance in a unified way. SOPs define specific procedures that employees must follow to ensure quality and consistency in the deliverables.
SOPs are the cornerstone of any quality system, providing clear and detailed guidelines for employees to follow. They are typically documented procedures that outline step-by-step instructions for performing specific tasks. SOPs may cover everything from manufacturing processes to customer service procedures, and they are critical for ensuring that everyone in the organization is on the same page.
Pharmacovigilance Systems
PV systems are designed to monitor, assess and make impact to the safety of medicinal products. It includes:
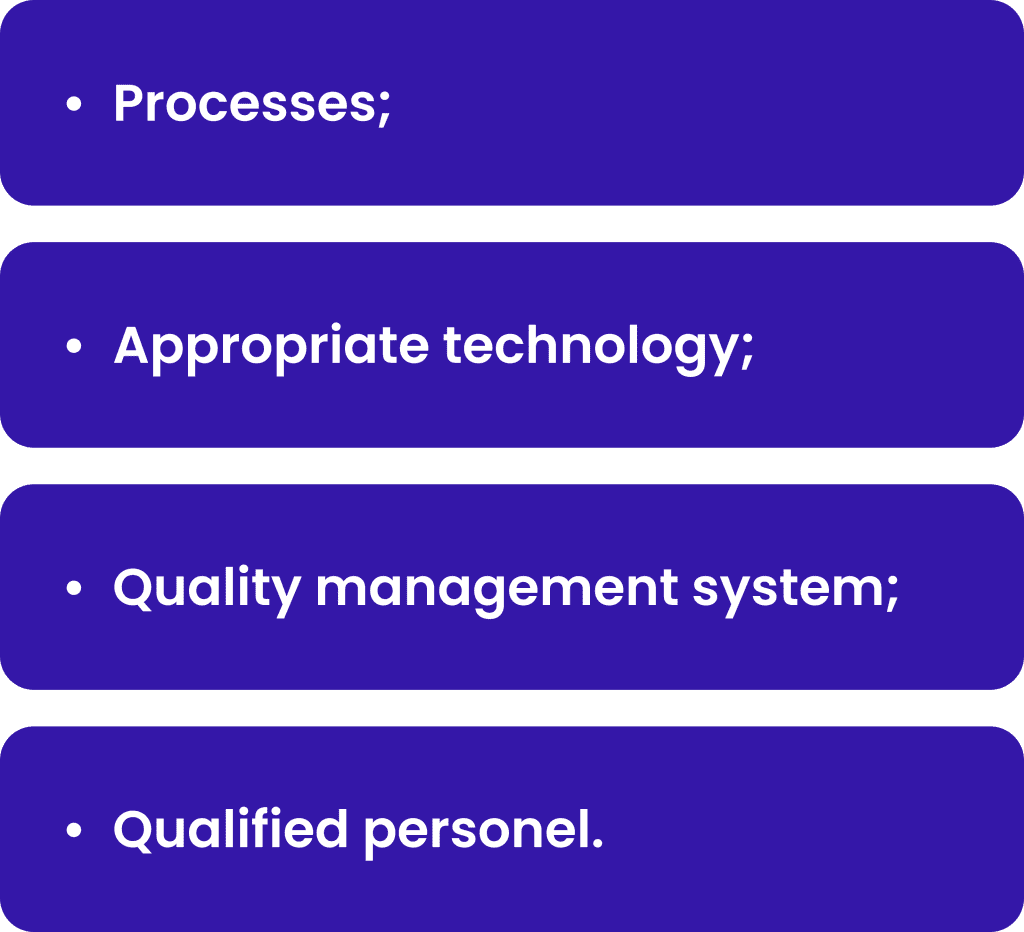
The processes should be compliant with the regulatory authorities requirements and should be designed to identify potential risks and mitigate them. The processes ensure the unified way of activities by the different persons through different territories. Appropriate technology must be implemented to support the processes and make it more efficient and traceable. The quality management system should be designed to ensure that the processes are controlled, monitored, and improved continuously. Overall qualified personnel shall perform processes, manage technology and ensure quality based on his/her responsibilities. Examples of each of these parts will be shortly described below.
PSMF
QMS and SOPs in place are not enough to be ready for PV system set up. Considering that each company has its own specific way of activities, its own communication pathways, its own partners and sources for safety information gathering, it is crucial to describe the exact company way of activities in one place. Such document is the Pharmacovigilance System Master File (PSMF), which outlines a company’s approach to continuous detecting, assessing and taking appropriate actions in respect of medicinal products. PSMF contains information:


The PSMF is a central repository of description of all pharmacovigilance-related activities, including policies and procedures, monitoring, and reporting of adverse drug reactions, and risk management systems. It is a crucial document for a MAH and regulatory authorities as it provides an overview of how the pharmacovigilance system of the company is organized. By providing PSMF, company state that company complies with regulatory requirements.
Pharmaceutical companies are required to maintain an updated PSMF routinely throughout the product lifecycle, starting with the preparation for submission of the first medicinal product until the expiration of the last batch of the last marketing authorization worldwide. The PSMF should reflect changes to the pharmacovigilance system, including changes in the company’s organization or structure, changes in pharmacovigilance activities, and changes in regulatory requirements as well as have an overview of pharmacovigilance system performance.
Risk Management Plan
All new applications for Marketing authorization of medicinal product must include a Risk Management Plan (RMP). The comprehensiveness of RMP depends on medicinal product itself and on selected application type of authorization. RMP should outline all safety information known for the time of medicinal product application, e.g. the identified risks, potential risks, and missing information such as drug exposure during pregnancy or usage by specific patient populations that have not been exposed to the product before. If some risks are known or potential one, RMP should also describe the risk management system, risk minimization measures, and their effectiveness measurement. Risk management system allow company to ensure public health by stating the current knowledge on specific medicine safety as well as stating how already known risks will be managed/mitigated.
The RMP must be updated when significant new information emerges, and the MAH may need to conduct post-marketing studies or maintain a safety information registry. Any major risks that affect the benefit-risk ratio may result in changes to the existing marketing authorization. Advanced Therapy Medicinal Products (ATMPs) derived from stem cells, tissue engineering, or gene therapy technologies require additional risk management considerations, which must be described in the RMP.
QPPV & Other Pharmacovigilance Personnel
Appointed Qualified Person for Pharmacovigilance (QPPV) is mandatory for all medicinal products authorized within European Union. The QPPV is a physical person, who has a legal liability against authorities, to ensure functioning Pharmacovigilance system, oversee its performance and data integrity. However, the final responsibility on safety of medicinal products remains on MAH. The EU-QPPV must reside in the European Economic Area (EEA) and ideally in the same country as PSMF or main PV activities are performed.
While EU-QPPV has a responsibility on functioning PV system, it can not be done only by himself/herself in most of the cases. Daily pharmacovigilance activities are done in two levels:


Local Pharmacovigilance Systems
Depending on company portfolio and type of medicines, it could be that two-three persons can fulfil requirements of global PV activities. However local PV system shall have persons, who can communicate in national language of the country, thus the number of personnel for local PV activities depends on number of countries of authorization.
The reliable and qualified network of local persons is one of the main key point of success to ensure pharmacovigilance activities compliance at country level and continuous improvement. Local persons hold expertise of local requirements, ensure compliance to it, and monitor any updates of legislation changes relevant to PV system. Local PV persons are the first contact point between regulatory authority and QPPV in any question. As a local experts, local PV persons are responsible to train other personnel of MAH about pharmacovigilance and shared reporting responsibility. So, local PV persons proactively can impact PV systems and mitigate some risks by providing expertise on specific questions to specific companies.
Most of the non-EU countries also require to have local qualified person responsible for pharmacovigilance, who would be the firstly accountable to national regulatory authority in respect of local PV activities and other local PV requirements.
Eudravigilance Profile
EudraVigilance (EV), central databased maintained by EMA, serves as a central repository not only for individual case safety reports (ICSRs), but also for administrative information such as the location of the Pharmacovigilance System Master File (PSMF), Qualified Person for Pharmacovigilance (QPPV), and Marketing Authorizations (MAs) of each Marketing Authorization Holder (MAH). Information existing in EV shall be up to date, so MAH is obliged to do updates in case of changes. All updates shall be done by properly qualified persons. MAHs are obligated to register for an EV profile or convert an existing clinical trial sponsor profile to a MAH profile. The QPPV or a trusted representative must attend one or more EV training sessions, which occur several times a year and have an attendance fee.
The PSMF appendices must include evidence of QPPV registration within EV. If a MAH outsources QPPV and EV profile management to an established pharmacovigilance provider, the required EV certifications are likely already held by employees, which can avoid unnecessary delays and costs. Required EV certifications has been identified as a significant issue by European Economic Area pharmacovigilance inspectors.
Safety Database & ADR Reporting System
Each MAH must have a properly organized systems for safety information (ICSRs, special situations, other safety information for interest) collection, processing, reporting, analysis. This is hardly could be done manually, so legislation compliant computerized system (Global safety database) is an essential tool, which help manage all safety information. Global safety database is designed to collect, store, manage, report and analyze data related to safety information, allowing organizations to identify potential safety concerns and take appropriate action.
Key aspects of safety databases and ADR reporting systems:

Currently all safety databases shall be compliant with the E2B R3 format to be able to report to Eudravigilance and fulfill the legal requirements. They help organizations to identify potential safety concerns associated with drugs and medical devices and take appropriate action to protect patients. Considering, that global safety database is an IT tool, which usually cost significantly, it is worth to consider that PV service provider already have global safety database and the cost would be shared between clients.
Literature Screening
MAH shall be proactive and actively seek to collect all available safety data with its’ medicines. One of the main sources of safety information is medicinal literature.
Medicinal literature shall be monitored on global and local activities. Global literature monitoring shall include well known scientific medicinal database. The main aim of global literature monitoring is to collect ICSRs and gather scientific knowledge on medicinal product. Local literature monitoring is usually done by the local person, because the medicinal journals are usually published in national language. Literature monitoring is PV activity, which require that sample of it would be quality checked by the second person.
Signal Management
Literature is one of the sources of knowledge sharing. MAH’s personnel who screens literature may be the one, who the first would notice unusual change of adverse events ratio, new adverse events or change risks of the medicinal product. In addition to such ad hoc notices, the routine process of signal detection shall be in place.
As per GVP Module IX, a signal refers to any information that arises from one or multiple sources, which includes observations and experiments indicating a potentially causal association or a new aspect of a known association between an intervention and an event or set of related events. This applies to adverse and beneficial outcomes deemed likely enough to warrant further investigation. Safety signals may originate from various sources such as safety and clinical trial databases, ICSRs, aggregate reviews, published literature, Competent Authorities, and manufacturing data.
Signal detection is the process of searching for and identifying signals using data from any available source. The methodology employed should consider the nature of the data, the type of medicinal product, and its characteristics. Depending on the data set size, signal detection may involve a review of ICSRs, statistical analyses, or both, and should be meticulously documented.
The management of signaling aims to identify risks earlier, define them more clearly, and communicate them more effectively. It is important to note that not all signals represent risks, and not all of them require additional regulatory action, such as an update of the Summary of Product Characteristics (SmPC), following an assessment. Nonetheless, the signaling process plays a crucial role in pharmacovigilance as it ensures the monitoring and control of potential risks.
Documentation & Storing
The golden rule in pharmacovigilance is: “If You have done some activity, You have to document it. If it is not documented, it was not done”. All activities personnel are performing shall be properly documented, that all steps would be traceable. All documentation of performed activities shall be properly stored and retrieved any time needed within 10 years after the expiration of the last MA of MAH. MAH shall consider how to set up computerized system, which would allow for data to be available only for relevant personnel, to be retrievable, ensure business continuity and avoid any data loss.
Summary
The EU framework for pharmacovigilance is comprehensive, but there are also specific national requirements to consider. Good Pharmacovigilance Practice modules guide meeting these requirements, although setting up a system can be complex, time-consuming, and costly. Many companies choose to outsource some or all their pharmacovigilance activities to third-party providers, which holds expertise on such activities. However, the marketing authorization holder remains ultimately responsible for product safety. The MAH needs to ensure that the provider’s systems and processes are compliant and validated and that their staff is qualified, experienced, and trained.
Here at Insuvia, we offer quality-driven pharmacovigilance services. For more information, contact us at [email protected].


