August 03, 2020
Bringing the Dossiers into Compliance in the EAEU
Until December 31, 2025, the Eurasian Economic Union (EAEU) which consists of Russia, Belarus, Kazakhstan, Kyrgyzstan, and Armenia shall form a single market for medicines. One of the most important milestones in the process of formation of a single pharmaceutical market of the Eurasian Economic Union (EAEU) is the deadline for bringing the Dossiers into compliance with the Eurasian Economic Union (EAEU) rules based on national provisions with the newly established requirements for demonstrating drug safety, efficacy, and quality.
The procedure for bringing the dossiers into compliance with the Eurasian Economic Union (EAEU) rules has raised many concerns among the pharmaceutical industry since it affects all medicinal products that were previously authorized among EAEU member states according to their national rules. The dossiers of such products shall either be brought to compliance with the new requirements or their authorizations will be terminated upon the deadline of December 31, 2025. Anticipating a large amount of work for all holders of registration certificates should already now begin to think out a strategy for alignment, work through the existing gaps, and think about the problem of missing preclinical and clinical data.
In this overview, we aim to raise awareness of the importance of this procedure and seek to provide key strategic considerations for our clients to ensure the proper marketing of their medicines in EAEU.
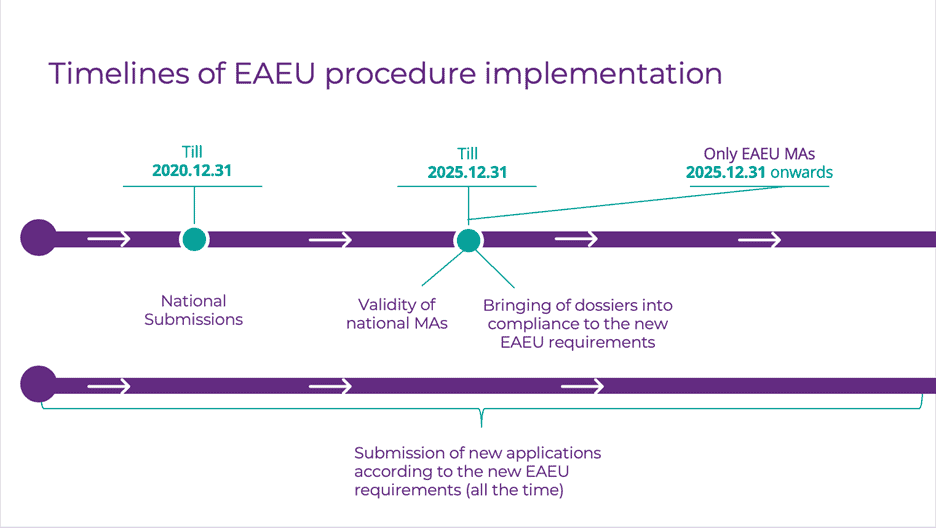
What are the timelines of EAEU procedure implementation and for bringing dossiers into compliance with Eurasian Economic Union (EAEU) rules?
- Submission of new applications according to the new EAEU requirements (all the time);
- National Submissions (till 31/Dec/2020);
- Bringing of dossiers into compliance to the new EAEU requirements (till 31/Dec/2025);
- Validity of national MAs (till 31/Dec/2025);
- Only EAEU MAs (31/Dec/2025 onwards).
How the procedure of bringing old dossiers into compliance with EAEU requirements looks like?
The procedure of bringing a dossier into compliance with the new requirements is similar to other registration procedures, such as initial registration, renewal, and a variation. It is performed similarly as a Mutual Recognition Procedure (MRP), although it differs from it in terms of timelines and scope of assessment. The process involves the selection of a Reference Member State (RMS) – if the medicine is registered in more than one member state of the EAEU before the entry into force of the Agreement1 or before 31 December 2020, the applicant should choose one of the countries to act as the RMS. The documentation flow between the RMS and Concerned Member States (CMS) is similar to the standard process of an MRP. However at the same time, there is a difference from the MRP: if the drug was registered according to national procedures in at least 3 member states of the Union within 5 years or more, the registration certificate issued after the procedure of Bringing the Dossier into Compliance with the EAEU rules will immediately have an indefinite validity.
As part of the procedure of bringing old dossiers into compliance with EAEU requirements, the RMS carries out a procedure for assessing the completeness of the dossier and its compliance with the application requirements. This is done within 14 business days and is called a “validation procedure”. If at the validation stage questions arise regarding the completeness of the dossier, the applicant is sent a request for the submission of the missing documents and data. This takes up to 90 calendar days. It should be noted that the deadline for the applicant to provide the missing data and documents, as in the MRP, is not extended.
Secondly, the RMS shall prepare a final assessment report on the efficacy, safety, and quality. The report shall become publicly available once the confidential information has been removed and the document has been entered in the Union Register of Medicinal Products. At this point, the work of the RMS is complete and the applicant is granted a registration certificate with the indicated RMS. The modules of the expert reports of the RMS and the modules of the registration dossier of the medicinal product shall be available in the recognition countries chosen by the applicant within 5 working days of the end of the work in the RMS.
In the next stage, the evaluators of the Concerned Member State (CMS) evaluate the reports submitted by the RMS and may, if necessary, request additional information from the applicant. A period of 90 days is given for this action, after which the following 10 days are allowed for the made decision approval. It is important to note that the CMS only reviews Module 1 and the assessment reports of the RMS, however, the CMS may check the compliance of the assessment reports of the RMS with Modules 2-5.
Following the assessment, the registration certificate shall be issued by the CMS under the same number as that issued by the RMS. In a situation where the applicant does not immediately identify all the CMSs, the applicant shall be entitled to list them at any time. In such a case, the MRP will start in the new Member States within 5 working days of the establishment of the updated list. Lastly, in order to carry out the renewal procedure simultaneously in all CMS, the registration certificate for medicinal products authorized in those Member States before 2020 December 31 is issued for the remaining period of validity of the registration certificate.
Aspects to be considered by MAHs
Aspects of the assessment of the whole dossier are provided in paragraphs 177-180 of the Union Registration Rules. Paragraph 178 is one of the most important because it provides important clarifications regarding the assessment of a medicinal product dossier and its bringing into compliance. In short, it states that a reassessment of the risk-benefit balance is not required when assessing the marketing authorization application dossier for a medicinal product within bringing the dossier into compliance with the requirements of the Union. In order to treat the information provided correctly, it would be rational to consider the following points:
- Examination of any drug reviews several types of evaluations and, based on a summary of their results, a final decision is usually made;
- The assessments performed during the assessment include safety, quality, administrative documentation, and performance evaluation;
- To bring the medicinal product into compliance, it is necessary to confirm the medicinal product’s safety and efficacy for each indication given in the summary of product characteristics (SmPC).
It is, therefore, necessary to provide a complete dossier to the Union in accordance with the requirements of Annex 1 to the Registration Rules, but it is not required to rewrite the dossier in cases where studies were performed prior to the introduction of good practice in the Regulation.
All facts considered, where to begin?
- Analyse your product portfolio. Check the registration status among the EAEU member states, including registered indications, strengths, forms and pack size. Furthermore, take in to account that EAEU rules give the regulators the opportunity to review not only safety, but also the effectiveness of a drug for one, several, or all indications.This may bring up the need to demonstrate effectiveness in additional clinical trials, and maybe particularly relevant for old drugs, the effectiveness of which, with many indications from the standpoint of current knowledge in the field of clinical medicine, is questioned;
- Choose the right Reference Member State. You may want to choose the Reference Member State (RMS) which has all the indications, strengths, forms, and pack sizes that you require to have in the EAEU marketing authorization. Many MAHs shall choose Russia as it is the largest and likely most important market to many. Also whilst choosing RMS you should consider thinking about Health Authorities with working national Scientific Advice function (Kazakhstan) or with advanced regulatory legislation (Russia, Kazakhstan, Belarus);
- Perform Dossier Gap analysis. We recommend performing gap analysis to assess the completeness and quality of the existing dossier. During this process, a dossier is reviewed and a list of missing documents and data is compiled (CTD availability, EAEU GMP certificate availability, need for additional clinical trials, module 3 correspondence to the required Pharmacopeia);
- Bringing the Dossier to EAEU requirements. After performing a dossier gap analysis and identifying which documents are missing, it is appropriate to develop a strategic plan to achieve regulatory compliance within the set deadlines. To fit into the set time intervals in your strategic plan, it is recommended to consider how much time will be needed and will be allocated to prepare the necessary missing documents and conduct medical writing. If there are difficulties and issues in the process of seeking to bring dossiers into compliance with EAEU requirements, it is appropriate to seek the scientific advice of Health Authorities (e.g. Kazakhstan).
Conclusion
Thus, all these described processes are necessary to agree to a purposeful and full-fledged regulatory cycle, as a result of which the authorized bodies receive only those medicines whose safety and efficacy will henceforth be demonstrated. These Union rules on the registration of medicinal products are also considered to be well prepared and balanced, as they allow marketing authorization holders to respond rationally and without haste to additional requirements and to adequately defend their interests through regulated dispute resolution procedures.
If you need additional information regarding bringing dossier into compliance with EAEU rules contact us!
Back to news