
We live in the interim period for Clinical Trial Regulation (CTR) coming fully into effect. Despite the fact that CTR entered into force back in 2014, its application was.limited, due to technical IT difficulties. 31st Jan 2022 was the go-live date for the EU Clinical Trial Database and Portal. It enabled the full application of CTR. 31st of January 2023 is the end of one timeline of the transition period for CTR within the EU. Then all initial applications for clinical trials to conduct in the EU/EEA must be submitted to Clinical Trial Information System (CTIS).
The main goal of CTIS – is to facilitate and make it more transparent the whole processes of.Clinical trial management for sponsors, as well as.regulators making all information exchange available in one place. EMA announced invitation and agenda for CTIS Sponsor user training program: Clinical Trials Information System (CTIS) sponsor end user training programme (europa.eu)
Taking this opportunity, we would like to remind you and highlight other upcoming changes, especially in safety reporting requirements. Let’s take a look at the comparison for safety reporting requirements:
Table 1. Safety reporting requirements for clinical trials and the post-authorization phase.
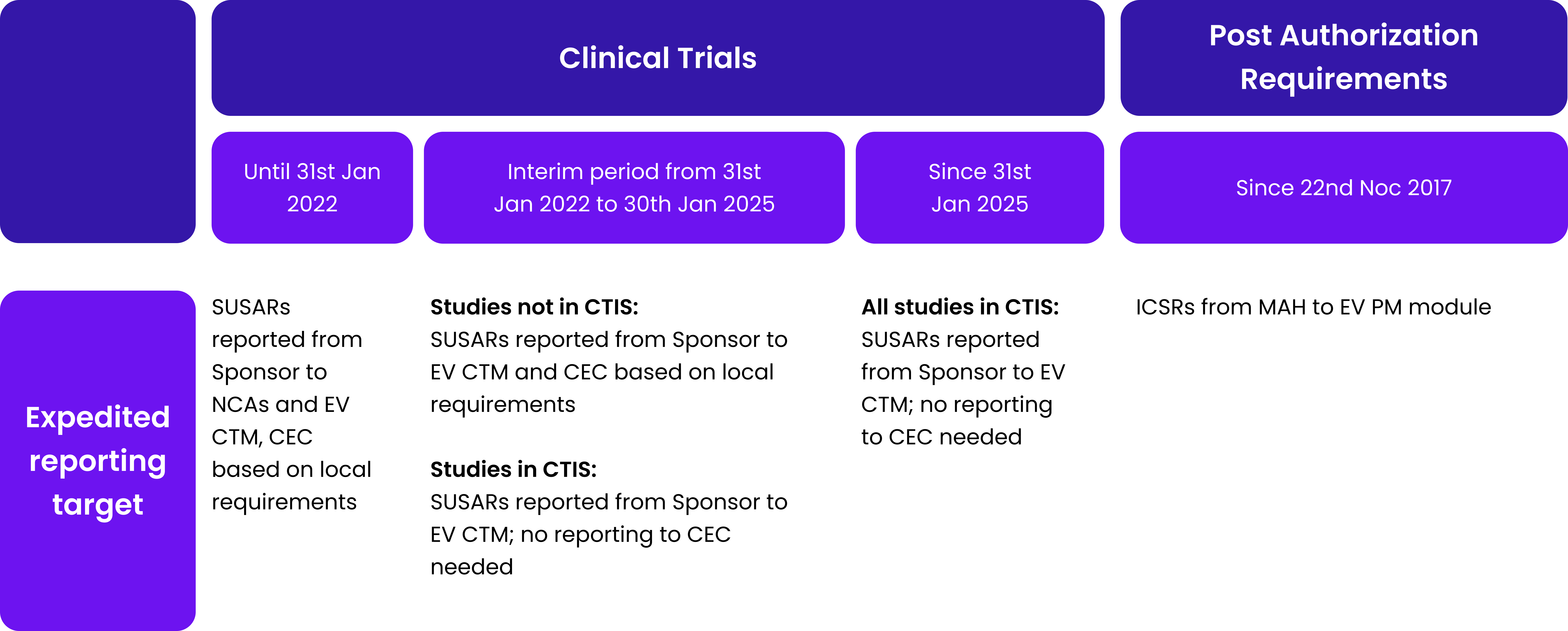


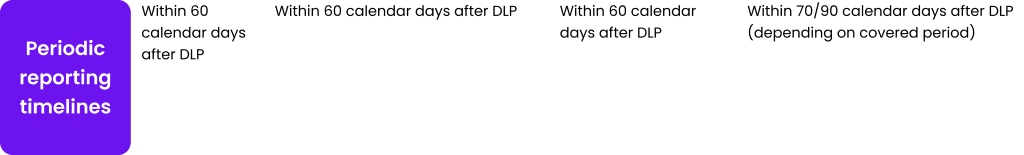
Abb: SUSAR – suspected unexpected serious adverse reaction; NCA – national competent authority; EV CTM – Eudravigilance Clinical trial module; CEC – concerned ethic committee; ICSR – individual case safety report; MAH-Marketing authorization holder; EV PM – Eudravigilance post marketing module; DSUR – development safety update report; IMP – investigational medicinal product; PSUR – periodic safety update report; DLP – data lock point.
As Table 1 shows, timelines for safety reporting remain unchanged. The introduction of CTIS changes only the target for submission of clinical trial documentation and make it.centralized – one point one time of submission, comparing to previous requirements to submit.the same information several times to separate stakeholders. It is easy to notice that safety reporting requirements for clinical trials are.made with the analogy to requirements in post authorization field.
On the other hand, EU regulators recognize that centralization may not be the best solution for every clinical trial. Specific situations may require decentralization. Recently, guideline (not legally binding) was published with recommendations on decentralized elements in clinical trials: Recommendation paper on decentralised elements in clinical trials (europa.eu)
Additionally, it is worth mentioning that ICH already published draft guidelines for Clinical electronic Structured Harmonized protocol (M11). Thus, there is a high possibility that after 31st of January 2025, we will have all studies in CTIS, harmonized structure of clinical trial protocol and simplified reporting system. Moreover, it will be comparable to post authorization system.
Considering that Clinical Trial Regulation shall.be followed as it is, without specific implementation into national law of Member States, it is.expected that harmonized requirements for whole Clinical Trial Management facilitate the processes and will allow to Sponsors keep higher quality of studies.
If you would like to know more information, please do not hesitate to contact us.
Related news
-
 12 March, 2026 Pharmacovigilance Articles
12 March, 2026 Pharmacovigilance ArticlesPharmacovigilance System Transfer: How to Manage It Without Losing Control
-
04 March, 2026 Regulatory Affairs Articles
Before You In-License: GMP Risks and Third-Country CMO Realities
-
27 February, 2026 Regulatory Affairs Articles
Labelling Exemptions and Foreign Packaging in the EU/EEA: A Practical Guide for Regulatory Strategy


