
IDMP standards implementation to product information management
EMA has embraced the challenge of being the first agency to implement the ISO standards for the identification of medicinal products (ISO IDMP). As core component of this process, the Product Management Service (PMS) will become the central database for product information across the European Union. This transition is a significant change to the pharmacovigilance industry and requires thorough preparation from various stakeholders, including major input from the Marketing Authorization Holders (MAHs).
The implementation of the new standards will bring notable benefits to pharmacovigilance: adverse event reports will be based on a harmonized set of product definitions, improving the quality of data used for signal management, speeding up communication, decision-making, and regulatory actions. The PMS system will also facilitate the reuse of product information in other regulatory and non-regulatory systems, including benefits for European citizens overall.
Integrating IDMP standards into PMS requires the commitment of MAHs to map a new set of data elements from the existing XEVMPD structure to the new system. In addition to the information currently mandated by XEVMPD, the following will be obligatory:
- Information about manufacturing operations, procedures, and authorized facilities for the active components, intermediates, and final products.
- Identification of the marketing status at the packaging level, indicating which products/packages are available in the EU market.
- Details regarding the forms and strengths of each medicinal product that can be administered.
- Description of the manufactured item that outlines the authorized pharmaceutical form of the product, and if applicable, its state before conversion into the administrable pharmaceutical form.
- Packaging details such as the container for the packaged item, quantity, and material for both the inner and outer packaging.
- Legal classification of supply (prescription or over-the-counter) at the product level if consistent across all packs; at the pack level if it varies.
- Information on the composition of medicinal products, including the source of ingredients, description of composition groups, and requirements for submitting details for products certified under TSE, ASMF, CEP, VAMF & PMF certifications.
Detailed guidance on the additional information that will be required in PMS, can be downloaded after filling the contact form at the bottom of this page.
PMS data elements
As part of ISO IDMP implementation process, product Management Service (PMS) will become the central database for product information management across the European Union. It will combine data from both the Extended EudraVigilance Medicinal Product Dictionary (XEVMPD), also known as the Article 57 database, and SIAMED, an internal EMA database that contains centrally authorized products. To help the MAHs navigate the PMS data and related business rules, EMA released extensive PMS implementation guide which consists of nine chapters, seven already published and two pending.
According to the PMS Implementation Guide Chapter 2 released by EMA, more than 150 data elements will be applicable in the PMS, belonging to 6 categories by type:
- Medicinal product;
- Marketing authorisation information;
- Therapeutic (product) indication;
- Packaged medicinal product;
- Ingredient;
- Pharmaceutical product.
Each data element was also classified by its conformance: mandatory, conditional and optional. More than half of PMS data fields are mandatory (Figure 1).
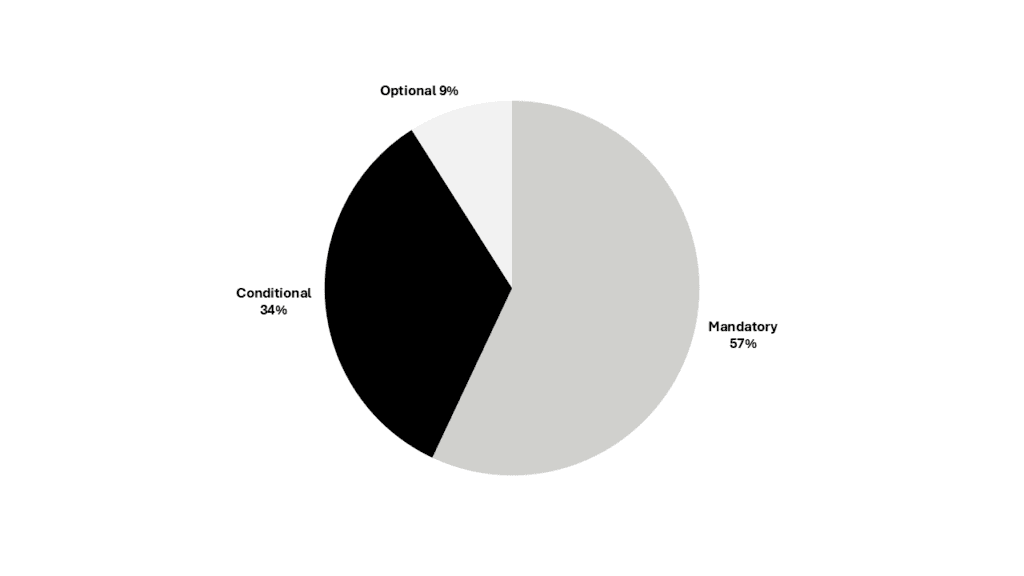
Figure 1. Data elements by conformance (click to enlarge)
Data migration to PMS
In the PMS Implementation Guide Chapter 7 released by EMA, each data element is classified by its origin. Almost a third of data elements lack migrated information (Figure 2).
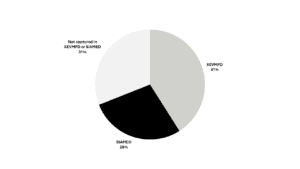
Figure 2. Data elements by migration source (click to enlarge)
For MAHs to also have a clear picture on data migration, EMA provides the summary of origins for different data element types. It demonstrates that certain data element types such as Ingredients, Route of administration, QPPV, PhV Contacts, Country/Languages, and Attachments derive from XEVMPD. Fields sourced from SIAMED include Cross Reference, Orphan Designations, Package Component, and Authorized Pharmaceutical Form.
However, there is a significant amount of data that does not derive from XEVMPD or SIAMED. For example, fields such as Shelf life/Storage, Data Carrier, Marketing Status, and Device lack migrated information. Data in some other fields partially derive from either XEVMPD or SIAMED, yet additional information may need to be extracted from other sources beyond these databases (Figure 3).
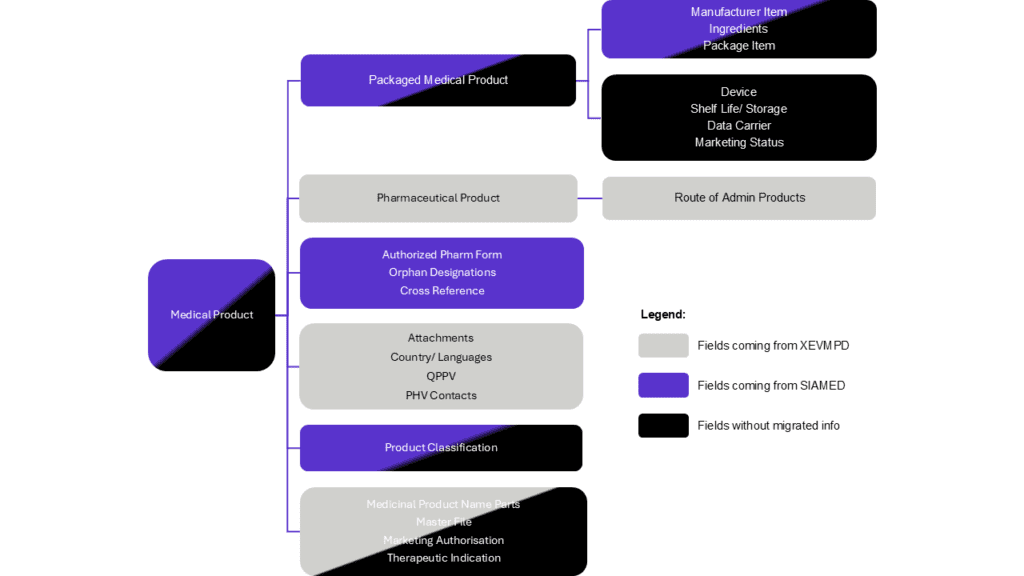
Figure 3. Overview of the origin of the product data in PMS (click to enlarge)
To summarize, among the must-fill data elements fields in the PMS, a total of 38 remain without migrated information and will not transition from SIAMED or XEVMPD. While some of these PMS fields, such as specific identifiers, are system-generated, the majority of them will need to be extracted by the MAHs from alternative sources. You can find the full list of these mandatory, not migrated data elements and their business rules can be downloaded after filling the contact form at the bottom of this page.
Are You already ready for PMS?
For quite a long time, all ISO IDMP implementation process was managed by EMA/NCAs and selected partners. MAHs were required to follow what is happening without active participation. However, one of the deadline for MAHs actions has just ended with the Q1 of 2024. Check here if You are ready for PMS: ISO IDMP: EMA‘s Recommendations for Data Migration – Insuvia
Conclusion and key takeaway
Embracing the ISO IDMP standards to product information management is a significant challenge for the pharmacovigilance industry, and demands careful planning and cooperation among various internal and external partners, including the MAHs. Understanding IDMP’s ultimate goal to streamline product information extraction for patient safety, it is essential for life sciences companies to adjust their preparations accordingly.


